86 Neurotransmitters and Receptors
Cholinergic Neurons and Receptors
Acetylcholine is a neurotransmitter in the central and peripheral nervous systems that affects plasticity, arousal, and reward.
Learning Objectives
Identify the cholinergic neurons and receptors in the autonomic system
Key Takeaways
Key Points
- The neurotransmitter acetylcholine (ACh) is the only neurotransmitter used in the motor division of the somatic nervous system and the principal neurotransmitter at autonomic ganglia.
- In the CNS, the neurons that release and respond to ACh comprise the cholinergic system, which causes anti-excitatory effects.
- ACh plays a role in synaptic plasticity, including learning and short-term memory.
- ACh may bind either muscarinic or nicotinic receptors.
- ACh is synthesized in cholinergic neurons (such as those in the nucleus basalis of Meynert) from choline and acetyl-CoA using an enzyme called choline acetyltransferase.
Key Terms
- choline acetyltransferase: Abbreviated as ChAT, this is an enzyme that is synthesized within the body of a neuron. It is then transferred to the nerve terminal via axoplasmic flow. The role of choline acetyltransferase is to join Acetyl-CoA to choline, resulting in the formation of the neurotransmitter acetylcholine.
- autonomic ganglia: Clusters of neuronal cell bodies and their dendrites that are a junction between the autonomic nerves originating from the central nervous system and the autonomic nerves innervating their target organs in the periphery.
- nicotinic receptors: Also called nAChRs, these are cholinergic receptors that form ligand-gated ion channels in the plasma membranes of certain neurons and on the postsynaptic side of the neuromuscular junction.
Acetylcholine
Acetylcholine (ACh) is an organic, polyatomic ion that acts as a neurotransmitter in both the peripheral nervous system (PNS) and central nervous system (CNS) in many organisms, including humans. Acetylcholine is one of many neurotransmitters in the autonomic nervous system (ANS) and the only neurotransmitter used in the motor division of the somatic nervous system (sensory neurons use glutamate and various peptides at their synapses ).
Acetylcholine is also the principal neurotransmitter in all autonomic ganglia. In cardiac tissue, acetylcholine neurotransmission has an inhibitory effect, which lowers heart rate. However, acetylcholine also behaves as an excitatory neurotransmitter at neuromuscular junctions in skeletal muscle.

Acetylcholine: The chemical structure of acetylcholine is depicted.
Acetylcholine was first identified in 1914 by Henry Hallett Dale for its actions on heart tissue. It was confirmed as a neurotransmitter by Otto Loewi, who initially gave it the name Vagusstoff because it was released from the vagus nerve. They jointly received the 1936 Nobel Prize in physiology or medicine for their work. Acetylcholine was also the first neurotransmitter to be identified.
Functions
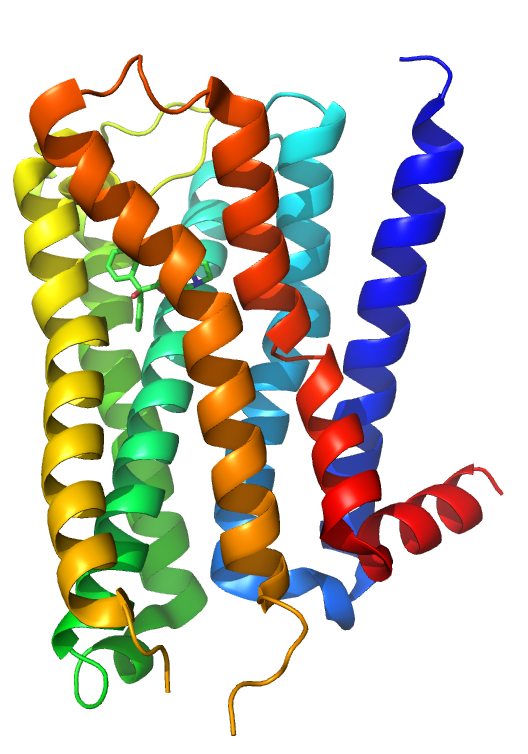
Muscarinic acetylcholine receptor M2: This human M2 muscarinic acetylcholine receptor is bound to an antagonist (ACh).
Acetylcholine has functions both in the peripheral nervous system (PNS) and in the central nervous system (CNS) as a neuromodulator. In the peripheral nervous system, acetylcholine activates muscles and is a major neurotransmitter in the autonomic nervous system. In the central nervous system, acetylcholine and its associated neurons form the cholinergic system.
When acetylcholine binds to acetylcholine receptors on skeletal muscle fibers, it opens ligand-gated sodium channels in the cell membrane. Sodium ions then enter the muscle cell, initiating a sequence of steps that finally produce muscle contraction. Although acetylcholine induces contraction of skeletal muscle, it acts via a different type of receptor to inhibit the contraction of cardiac muscle fibers.
In the autonomic nervous system, acetylcholine is released in the following sites: all pre- and post-ganglionic parasympathetic neurons, all pre-ganglionic sympathetic neurons, some post-ganglionic sympathetic fibers, and in the pseudomotor neurons to sweat glands.
In the central nervous system, ACh has a variety of effects as a neuromodulator for plasticity, arousal, and reward. ACh has an important role in the enhancement of sensory perceptions when we wake up and in sustaining attention.
Damage to the cholinergic (acetylcholine-producing) system in the brain has been shown to be plausibly associated with the memory deficits associated with Alzheimer’s disease. ACh has also been shown to promote REM sleep.
In the cerebral cortex, tonic ACh inhibits layer 4 neurons, the main targets of thalamocortical inputs while exciting pyramidal cells in layers 2/3 and 5. This filters out weak sensory inputs in layer 4 and amplifies inputs that reach the layers 2/3 and layer 5 excitatory microcircuits.
As a result, these layer-specific effects of ACh might function to improve the signal-to-noise ratio of cortical processing. At the same time, acetylcholine acts through nicotinic receptors to excite certain groups of inhibitory interneurons in the cortex that further dampen cortical activity.
One well-supported function of ACh in the cortex is an increased responsiveness to sensory stimuli, a form of attention. Phasic increases of ACh during visual, auditory, and somatosensory stimulus presentations have been found to increase the firing rate of neurons in the corresponding primary sensory cortices.
When cholinergic neurons in the basal forebrain are lesioned, animals’ ability to detect visual signals was robustly and persistently impaired. In that same study, an animals’ ability to correctly reject non-target trials was not impaired, further supporting the interpretation that phasic ACh facilitates responsiveness to stimuli.
ACh has been implicated in reporting expected uncertainty in the environment, based both on the suggested functions listed above and results recorded while subjects perform a behavioral cuing task. Reaction time differences between correctly cued trials and incorrectly cued trials, called the cue validity, was found to vary inversely with ACh levels in primates with pharmacologically and surgically altered levels of ACh. The result was also found in Alzheimer’s disease patients and smokers after nicotine (an ACh agonist) consumption.
Creation of ACh
Acetylcholine is synthesized in certain neurons by the enzyme choline acetyltransferase from the compounds choline and acetyl-CoA. Cholinergic neurons are capable of producing ACh.
An example of a central cholinergic area is the nucleus basalis of Meynert in the basal forebrain. The enzyme acetylcholinesterase converts acetylcholine into the inactive metabolites choline and acetate. This enzyme is abundant in the synaptic cleft, and its role in rapidly clearing free acetylcholine from the synapse is essential for proper muscle function.
Certain neurotoxins work by inhibiting acetylcholinesterase, leading to excess acetylcholine at the neuromuscular junction. This results in paralysis of the muscles needed for breathing and stops the beating of the heart.
Adrenergic Neurons and Receptors
Adrenergic receptors are molecules that bind catecholamines. Their activation leads to overall stimulatory and sympathomimetic responses.
Learning Objectives
Describe adrenergic neurons and receptors in the autonomic nervous system
Key Takeaways
Key Points
- Adrenergic receptors consist of two main groups, α and β, multiple subgroups (α1, α2, β1, β2, β3), and several subtypes of the α2 subgroup (α2A, α2B, α2C).
- Epinephrine binds both α and β adrenergic receptors to cause vasoconstriction and vasodilation.
- When activated, the α1 receptor triggers smooth muscle contraction in blood vessels in the skin, gastrointestinal tract, kidney, and brain, among other areas.
- When activated, the α2 receptor triggers inhibition of insulin and the induction of glucagon release in the pancreas, contraction of GI tract sphincters, and increased thrombocyte aggregation.
- When activated, the α2 receptor triggers inhibition of insulin and induction of glucagon release in the pancreas, contraction of GI tract sphincters, and increased thrombocyte aggregation.
Key Terms
- adrenoreceptor: These are a class of G protein-coupled receptors that are targets of the catecholamines, especially norepinephrine (noradrenaline) and epinephrine (adrenaline). Many cells possess these receptors, and the binding of a catecholamine to the receptor will generally stimulate the sympathetic nervous system.
- G protein-coupled receptors: These comprise a large protein family of transmembrane receptors that sense molecules outside the cell and activate inside signal transduction pathways and, ultimately, cellular responses. Any adrenergic effects on cells are generally mediated by G protein-coupled receptors.
- adrenergic receptor: Any of several sites in the surface membranes of cells innervated by adrenergic neurons.
The adrenergic receptors (or adrenoceptors) are a class of metabotropic G protein -coupled receptors that are targets of the catecholamines, especially norepinephrine or noradrenaline, and epinephrine ( adrenaline ). Although dopamine is a catecholamine, its receptors are in a different category.
Many cells possess these receptors, and the binding of an agonist will generally cause a sympathetic (or sympathomimetic) response (e.g., the fight-or-flight response). For instance, the heart rate will increase, pupils will dilate, energy will be mobilized, and blood flow will be diverted from non-essential organs to skeletal muscle.

Adrenaline (epinephrine): The 2D structure of adrenaline (epinephrine) is illustrated.

Noradrenaline (norepinephrine): The 2D structure of noradrenaline (norepinephrine) is illustrated here.
There are two main groups of adrenergic receptors, α and β, with several subtypes. α receptors have the subtypes α1 (a Gq coupled receptor) and α2 (a Gi coupled receptor). Phenylephrine is a selective agonist of the α receptor.
β-receptors have the subtypes β1, β2, and β3. All three are linked to Gs proteins (although β2 also couples to Gi), which in turn are linked to adenylate cyclase. Agonist binding thus causes a rise in the intracellular concentration of the second messenger cAMP. Downstream effectors of cAMP include the cAMP-dependent protein, kinase (PKA), which mediates some of the intracellular events following hormone binding. Isoprenaline is a nonselective agonist.
Adrenaline or noradrenaline are receptor ligands to α1, α2, or β-adrenergic receptors (the pathway is shown in the following diagram).
- α1 couples to Gq, which results in increased intracellular Ca2+ that results in smooth muscle contraction.
- α2, on the other hand, couples to Gi, which causes a decrease of cAMP activity, that results in smooth muscle contraction.
- β receptors couple to Gs, and increases intracellular cAMP activity, resulting in heart muscle contraction, smooth muscle relaxation, and glycogenolysis.
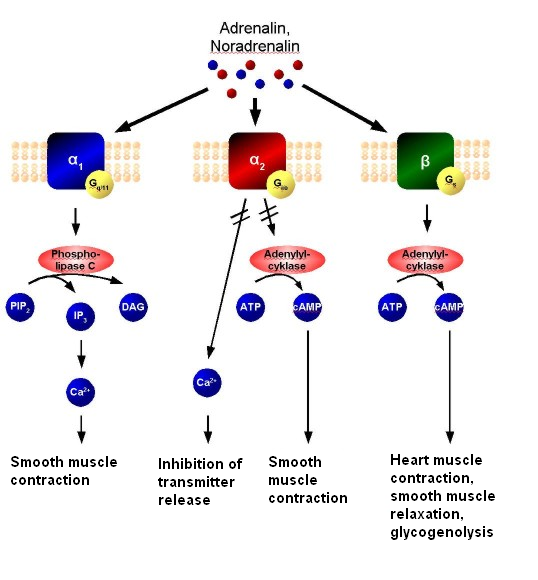
Adrenergic signal transduction: This schematic shows the mechanism of adrenergic receptors. Adrenaline and noradrenaline are ligands to α1, α2, or β-adrenergic receptors. α1-receptors couple to Gq, resulting in increased intracellular Ca2+ and causing smooth muscle contraction. α2 receptors couple to Gi, causing a decrease in cAMP activity and resulting in smooth muscle contraction. β-receptors couple to Gs, increasing intracellular cAMP activity and resulting in heart muscle contraction, smooth muscle relaxation, and glycogenolysis.
Adrenaline (epinephrine) reacts with both α- and β-adrenoceptors, causing vasoconstriction and vasodilation, respectively. Although α receptors are less sensitive to epinephrine, when activated, they override the vasodilation mediated by β-adrenoceptors. The result is that high levels of circulating epinephrine cause vasoconstriction. At lower levels of circulating epinephrine, β-adrenoceptor stimulation dominates, producing an overall vasodilation.
Smooth muscle behavior is variable depending on anatomical location. One important note is the differential effects of increased cAMP in smooth muscle compared to cardiac muscle. Increased cAMP will promote relaxation in smooth muscle, while promoting increased contractility and pulse rate in cardiac muscle.
α-receptors have several functions in common, but also individual effects. Common (or still unspecified) effects include: vasoconstriction of cardiac arteries (coronary artery), vasoconstriction of veins, and decreased motility of smooth muscle in the gastrointestinal tract.
α1-adrenergic receptors are members of the G protein-coupled receptor superfamily. On activation, a heterotrimeric G protein, Gq, activates phospholipase C (PLC).
The PLC cleaves phosphatidylinositol 4,5-bisphosphate (PIP2), which in turn causes an increase in inositol triphosphate (IP3) and diacylglycerol (DAG). The former interacts with calcium channels of the endoplasmic and sarcoplasmic reticulum, thus changing the calcium content in a cell. This triggers all other effects.
Specific actions of the α1-receptor mainly involve smooth muscle contraction. It causes vasoconstriction in many blood vessels, including those of the skin, gastrointestinal system, kidney (renal artery), and brain. Other areas of smooth muscle contraction are:
- Ureter.
- Vas deferens.
- Hair (arrector pili muscles).
- Uterus (when pregnant).
- Urethral sphincter.
- Bronchioles (although minor to the relaxing effect of β2 receptor on bronchioles).
- Blood vessels of ciliary body (stimulation causes mydriasis).
Further effects include glycogenolysis and gluconeogenesis from adipose tissue and the liver, as well as secretion from sweat glands, and Na+ reabsorption from kidney. Antagonists may be used in hypertension.
There are 3 highly homologous subtypes of α2 receptors: α2A, α2Β, and α2C.
α2-Receptor Effects
- Inhibition of insulin release in the pancreas.
- Induction of glucagon release from the pancreas.
- Contraction of sphincters of the gastrointestinal tract.
- Negative feedback in the neuronal synapses—presynaptic inhibition of noradrenalin release in CNS.
β1-Receptor Effects
- Increases cardiac output, by raising heart rate (positive chronotropic effect), increasing impulse conduction (positive dromotropic effect), and increasing contraction (positive inotropic effect), thus increasing the volume expelled with each beat (increased ejection fraction).
- Increases renin secretion from the juxtaglomerular cells of the kidney.
- Increases ghrelin secretion from the stomach.
β2-Receptor Effects
- Smooths muscle relaxation, e.g., in bronchi and the GI tract (decreased motility).
- Lipolysis in adipose tissue.
- Anabolism in skeletal muscle.
- Relaxes a non-pregnant uterus.
- Dilates arteries to skeletal muscle.
- Glycogenolysis and gluconeogenesis.
- Stimulates insulin secretion.
- Contracts the sphincters of the GI tract.
- Thickens secretions from the salivary glands.
- Inhibits histamine release from mast cells.
- Increases renin secretion from the kidney.
- Relaxation of bronchioles (salbutamol, a beta-2 agonist, relieves bronchiole constriction).
Agonists, Antagonists, and Drugs
Drugs effecting cholinergic neurotransmission may block, hinder, or mimic the action of acetylcholine and alter post-synaptic transmission.
Learning Objectives
Distinguish between the effects of an agonist versus an antagonist in the autonomic nervous system
Key Takeaways
Key Points
- Acetylcholine receptor agonists and antagonists have either direct effects on the receptors or act indirectly by affecting the enzyme acetylcholinesterase.
- Agents targeting ACh receptors may target either the nicotinic or muscarinic receptors for ACh.
- Atropine, an antagonist for muscarinic ACh receptors, lowers the parasympathetic activity of muscles and glands in the parasympathetic nervous system.
- Neostigmine is an indirect ACh receptor agonist that inhibits acetylcholinesterase, preventing the breakdown of acetylcholine. It is used in the treatment of myasthenia gravis and to reverse the effects of neuromuscular blockers used for anesthesia.
- Phenylephrine, marketed as a substitute for Sudafed for decongestant purposes, is an α1- adrenergic receptor agonist.
- Beta-blockers, as their name suggests, block the action of epinephrine and norepinephrine on β-adrenergic receptors and are used for the management of cardiac arrhythmias, cardio-protection after a heart attack, and hypertension.
Key Terms
- acetylcholinesterase: An enzyme that catalyzes the breakdown of the neurotransmitter acetylcholine.
- beta-blockers: Also called beta-adrenergic blocking agents, beta-adrenergic antagonists, beta-adrenoreceptor antagonists, or beta antagonists, these are a class of drugs used for various indications. As beta-adrenergic receptor antagonists, they diminish the effects of epinephrine (adrenaline) and other stress hormones.
- atropine: An alkaloid extracted from the plant deadly nightshade (Atropa belladonna) and other sources. It is used as a drug in medicine for its paralytic effects (e.g., in surgery to relax muscles, in dentistry to dry the mouth, in ophthalmology to dilate the pupils), though overdoses are fatal.
Blocking, hindering, or mimicking the action of acetylcholine has many uses in medicine. Drugs that act on the acetylcholine system are either agonists to the receptors that stimulate the system, or antagonists that inhibit it.
Acetylcholine receptor agonists and antagonists can have a direct effect on the receptors or exert their effects indirectly. For example, by affecting the enzyme acetylcholinesterase the receptor ligand is degraded. Agonists increase the level of receptor activation, antagonists reduce it.
Acetylcholine in the ANS
The vagus (parasympathetic) nerves that innervate the heart release acetylcholine (ACh) as their primary neurotransmitter. ACh binds to muscarinic receptors (M2) that are found principally on cells comprising the sinoatrial (SA) and atrioventricular (AV) nodes.
Muscarinic receptors are coupled to the Gi-protein; therefore, vagal activation decreases cAMP. Gi-protein activation also leads to the activation of KACh channels that increase potassium efflux and hyperpolarizes the cells.
Increases in vagal activity to the SA node decreases the firing rate of the pacemaker cells by decreasing the slope of the pacemaker potential and decreasing heart rate. By hyperpolarizing the cells, vagal activation increases the cell’s threshold for firing, which contributes to the reduction the firing rate.
Similar electrophysiological effects also occur at the atrioventricular AV node. However, in this tissue, these changes are manifested as a reduction in impulse conduction velocity through the AV node. In the resting state, there is a large degree of vagal tone on the heart, which is responsible for low, resting heart rates.
There is also some vagal innervation of the atrial muscle, and to a much lesser extent, the ventricular muscle. Vagus activation, therefore, results in modest reductions in atrial contractility (inotropy) and even smaller decreases in ventricular contractility.
Muscarinic Antagonists

Atropine: The 2D chemical structure of atropine is illustrated here.
Muscarinic receptor antagonists bind to muscarinic receptors, thereby preventing ACh from binding to and activating the receptor. By blocking the actions of ACh, muscarinic receptor antagonists very effectively block the effects of vagal nerve activity on the heart. By doing so, they increase heart rate and conduction velocity.
Atropine is a naturally occurring tropane alkaloid extracted from deadly nightshade (Atropa belladonna), Jimson weed (Datura stramonium), mandrake (Mandragora officinarum), and other plants of the family Solanaceae. Atropine’s pharmacological effects are due to its ability to bind to muscarinic acetylcholine receptors. It is an anti-muscarinic agent.
Working as a nonselective muscarinic acetylcholinergic antagonist, atropine increases firing of the sinoatrial node (SA) and conduction through the atrioventricular node (AV) of the heart, opposes the actions of the vagus nerve, blocks acetylcholine receptor sites, and decreases bronchial secretions. In overdoses, atropine is poisonous.
Nicotinic Agonists
A nicotinic agonist is a drug that mimics, in one way or another, the action of acetylcholine (ACh) at nicotinic acetylcholine receptors (nAChRs). Nicotinic acetylcholine receptors are receptors found in the central nervous system, the peripheral nervous systems, and skeletal muscles.
They are ligand-gated ion channels with binding sites for acetylcholine as well as other agonists. When agonists bind to a receptor it stabilizes the open state of the ion channel allowing an influx of cations.
The development of nicotinic acetylcholine receptor agonists began in the early nineties after the discovery of nicotine’s positive effects on animal memory. Nicotinic antagonists are mainly used for peripheral muscle paralysis in surgery, the classical agent of this type being tubocurarine, but some centrally acting compounds such as bupropion, mecamylamine, and 18-methoxycoronaridine block nicotinic acetylcholine receptors in the brain and have been proposed for treating drug addiction.
The nicotinic acetylcholine receptor agonists are gaining increasing attention as drug candidates for multiple central nervous system disorders such as Alzheimer’s disease, schizophrenia, attention-deficit hyperactivity disorder (ADHD), and nicotine addiction. In 2009 there were at least five drugs on the market that affect the nicotinic acetylcholine receptors.
Most indirect-acting ACh receptor agonists work by inhibiting the enzyme acetylcholinesterase. The resulting accumulation of acetylcholine causes a continuous stimulation of the muscles, glands, and central nervous system.
They are examples of enzyme inhibitors, and increase the action of acetylcholine by delaying degradation; some have been used as nerve agents (sarin and VX nerve gas) or pesticides (organophosphates and the carbamates). In clinical use, they are administered to reverse the action of muscle relaxants, to treat myasthenia gravis, and to treat symptoms of Alzheimer’s disease (rivastigmine increases cholinergic activity in the brain).
Beta Receptor Antagonists
Beta blockers (sometimes written as β-blockers) or beta-adrenergic blocking agents, beta-adrenergic antagonists, beta-adrenoreceptor antagonists, or beta antagonists, are a class of drugs used for various indications. They are particularly used for the management of cardiac arrhythmias, cardiac protection after myocardial infarction (heart attack), and hypertension.
As beta-adrenergic receptor antagonists, they diminish the effects of epinephrine (adrenaline) and other stress hormones. Beta blockers block the action of endogenous catecholamines —epinephrine (adrenaline) and norepinephrine (noradrenaline) in particular—on β-adrenergic receptors, part of the sympathetic nervous system that mediates the fight-or-flight response.
